Hemofilia merupakan salah satu penyakit turunan dimana darah sangat sukar membeku. Apa yang dimaksud dengan Hemofilia (hemophilia) ? Bagaimana patofisiologi hemophilia sehingga darah menjadi sukar beku?
Hemofilia merupakan salah satu penyakit turunan dimana darah sangat sukar membeku. Apa yang dimaksud dengan Hemofilia (hemophilia) ? Bagaimana patofisiologi hemophilia sehingga darah menjadi sukar beku?
Hemofilia adalah gangguan produksi faktor pembekuan yang diturunkan, berasal dari bahasa Yunani, yaitu haima yang artinya darah dan philein yang artinya mencintai atau suka. Walaupun sebenarnya maknanya tidak sesuai, namun kata hemofilia tetap dipakai.
Kelainan perdarahan yang diturunkan pertama kali didokumentasikan di abad kedua oleh Kerajaan Babilonia. Namun baru pada abad ke 18 dilaporkan adanya kemungkinan basis genetik untuk kelainan perdarahan ini dan mulai tahun 1950an transfusi fresh frozen plasma (FFP) digunakan. Pada tahun 1980an teknik rekombinan DNA untuk menproduksi faktor VIII (F VIII) dan faktor IX (F IX) mulai diterapkan.
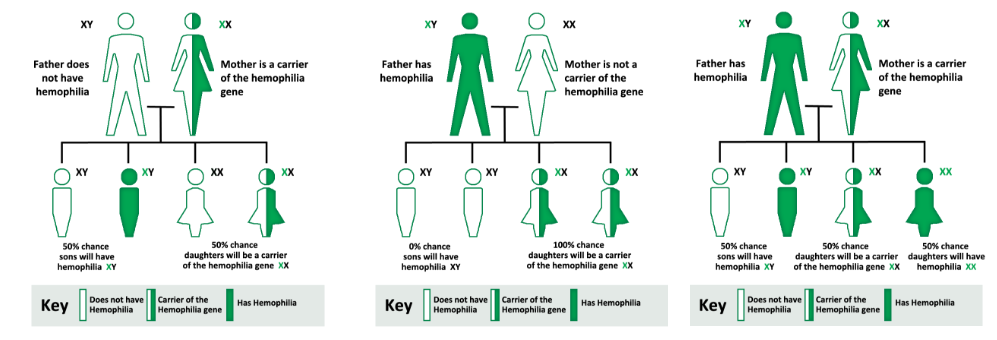
Hemofilia merupakan penyakit genetik yang diturunkan secara x-linked resesif berdasarkan hukum Mendel dari orang tua kepada anak-anaknya. Penyakit ini terjadi akibat kelainan sintesis salah satu faktor pembekuan, dimana pada hemofilia A terjadi kekurangan F VIII (Antihemophilic factor), sedangkan pada hemofilia B terjadi kekurangan F IX (Christmas factor). Hemofilia A mencakup 80-85% dari keseluruhan penderita hemofilia.
Secara klinis hemofilia dapat dibagi menjadi hemofilia ringan, hemofilia sedang dan hemofilia berat berdasarkan derajat kekurangan faktor pembekuan yang bersangkutan.
Hemofilia tersebar di seluruh ras di dunia dengan prevalensi sekitar 1 dalam 10 000 penduduk untuk hemofilia A dan 1 dalam 50 000 penduduk untuk hemofilia B.
Berdasarkan survei yang dilakukan oleh World Federation of Hemophilia (WFH) pada tahun 2010, terdapat 257 182 penderita kelainan perdarahan di seluruh dunia, di antaranya dijumpai 125 049 penderita hemofilia A dan 25 160 penderita hemofilia B.
Penderita hemofilia mencakup 63% seluruh penderita dengan kelainan perdarahan. Penyakit von Willebrand merupakan jenis kelainan perdarahan yang kedua terbanyak dalam survei ini setelah hemofilia yaitu sebesar 39.9%.
Di Indonesia, berdasarkan survei tersebut di atas, terdapat 334 orang penderita hemofilia A, 48 orang penderita hemofilia B dan 1006 orang penderita hemofilia yang belum ditentukan jenisnya.
Proses hemostasis tergantung pada faktor koagulasi, trombosit dan pembuluh darah. Mekanisme hemostasis terdiri dari respons pembuluh darah, adesi trombosit, agregasi trombosit, pembentukan bekuan darah, stabilisasi bekuan darah, pembatasan bekuan darah pada tempat cedera oleh regulasi antikoagulan, dan pemulihan aliran darah melalui proses fibrinolisis dan penyembuhan pembuluh darah.
Cedera pada pembuluh darah akan menyebabkan vasokonstriksi pembuluh darah dan terpaparnya darah terhadap matriks subendotelial. Faktor von Willebrand (vWF) akan teraktifasi dan diikuti adesi trombosit. Setelah proses ini, adenosine diphosphatase, tromboxane A2 dan protein lain trombosit dilepaskan granul yang berada di dalam trombosit dan menyebabkan agregasi trombosit dan perekrutan trombosit lebih lanjut. Cedera pada pembuluh darah juga melepaskan tissue factor dan mengubah permukaan pembuluh darah, sehingga memulai kaskade pembekuan darah dan menghasilkan fibrin. Selanjutnya bekuan fibrin dan trombosit ini akan distabilkan oleh faktor XIII.
Kaskade pembekuan darah klasik diajukan oleh Davie dan Ratnoff pada tahun 1950an dapat dilihat pada Gambar Kaskade pembekuan darah. Kaskade ini menggambarkan jalur intrinsik dan ekstrinsik pembentukan thrombin. Meskipun memiliki beberapa kelemahan, kaskade ini masih dipakai untuk menerangkan uji koagulasi yang lazim dipakai dalam praktek sehari-hari.
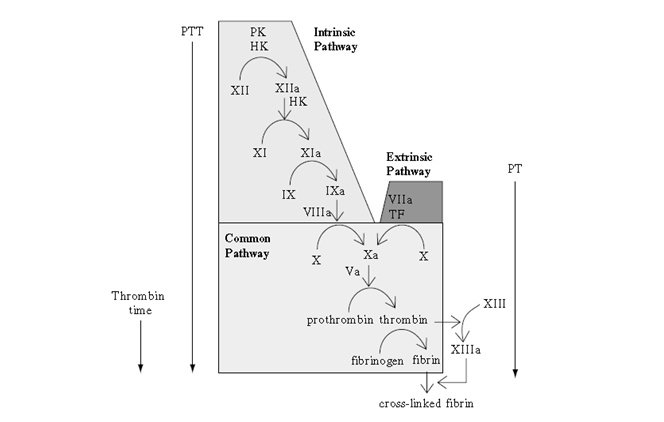
Gambar Kaskade pembekuan darah
Keterangan :
PK: Prekallikrein, HK: High molecular weight kininogen, TF: Tissue factor, PTT: Partial Prothrombin time, PT: Prothrombin time
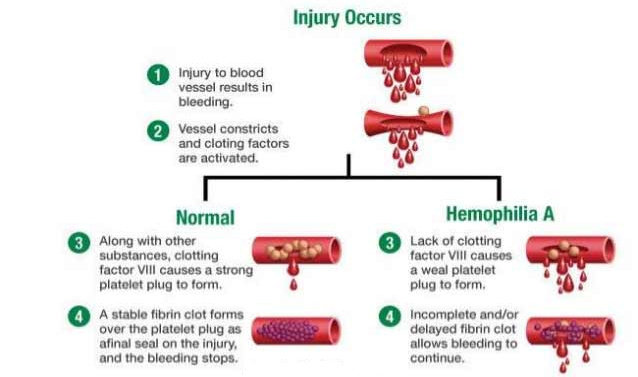
Pada penderita hemofilia dimana terjadi defisit F VIII atau F IX maka pembentukan bekuan darah terlambat dan tidak stabil. Oleh karena itu penderita hemofilia tidak berdarah lebih cepat, hanya perdarahan sulit berhenti. Pada perdarahan dalam ruang tertutup seperti dalam sendi, proses perdarahan terhenti akibat efek tamponade. Namun pada luka yang terbuka dimana efek tamponade tidak ada, perdarahan masif dapat terjadi. Bekuan darah yang terbentuk tidak kuat dan perdarahan ulang dapat terjadi akibat proses fibrinolisis alami atau trauma ringan.
Defisit F VIII dan F IX ini disebabkan oleh mutasi pada gen F8 dan F9. Gen F8 terletak di bagian lengan panjang kromosom X di regio Xq28, sedangkan gen F9 terletak di regio Xq27.2,14 Terdapat lebih dari 2500 jenis mutasi yang dapat terjadi, namun inversi 22 dari gen F8 merupakan mutasi yang paling banyak ditemukan yaitu sekitar 50% penderita hemofilia A yang berat. Mutasi gen F8 dan F9 ini diturunkan secara x-linked resesif sehingga anak laki-laki atau kaum pria dari pihak ibu yang menderita kelainan ini. Pada sepertiga kasus mutasi spontan dapat terjadi sehingga tidak dijumpai adanya riwayat keluarga penderita hemofilia pada kasus demikian.
Wanita pembawa sifat hemofilia dapat juga menderita gejala perdarahan walaupun biasanya ringan. Sebuah studi di Amerika Serikat menemukan bahwa 5 di antara 55 orang penderita hemofilia ringan adalah wanita.
Manifestasi klinis hemofilia A serupa dengan hemofilia B yaitu perdarahan yang sukar berhenti. Secara klinis hemofilia dapat dibagi menjadi hemofilia ringan (konsentrasi FVIII dan F IX 0.05-0.4 IU/mL atau 5-40%), hemofilia sedang (konsentrasi FVIII dan F IX 0.01-0.5 IU/mL atau 1-5%) dan hemofilia berat (konsentrasi FVIII dan F IX di bawah 0.01 IU/mL atau di bawah 1%)
Pada penderita hemofilia ringan perdarahan spontan jarang terjadi dan perdarahan terjadi setelah trauma berat atau operasi,. Pada hemofilia sedang, perdarahan spontan dapat terjadi atau dengan trauma ringan. Sedangkan pada hemofilia berat perdarahan spontan sering terjadi dengan perdarahan ke dalam sendi, otot dan organ dalam.
Perdarahan dapat mulai terjadi semasa janin atau pada proses persalinan. Umumnya penderita hemofilia berat perdarahan sudah mulai terjadi pada usia di bawah 1 tahun. Perdarahan dapat terjadi di mukosa mulut, gusi, hidung, saluran kemih, sendi lutut, pergelangan kaki dan siku tangan, otot iliospoas, betis dan lengan bawah. Perdarahan di dalam otak, leher atau tenggorokan dan saluran cerna yang masif dapat mengancam jiwa.
Diagnosis ditegakkan dengan anamesis, pemeriksaan fisik dan laboratorium. Anamnesis diarahkan pada riwayat mudah timbul lebam sejak usia dini, perdarahan yang sukar berhenti setelah suatu tindakan, trauma ringan atau spontan, atau perdarahan sendi dan otot. Riwayat keluarga dengan gangguan perdarahan terutama saudara laki-laki atau dari pihak ibu juga mendukung ke arah hemofilia.
Hasil pemeriksaan darah rutin dan hemostasis sederhana sama pada hemofilia A dan B. Darah rutin biasanya normal, sedangkan masa pembekuan dan masa thromboplastin parsial teraktifkan (APTT) memanjang, dan masa pembekuan thromboplastin abnormal. Masa perdarahan dan masa prothrombin (PT) umumnya normal.
Diagnosis pasti ditegakkan dengan memeriksa kadar F VIII untuk hemofilia A dan F IX untuk hemofilia B, dimana kedua faktor tersebut di bawah normal. Pemeriksaan petanda gen hemofilia pada kromosom X juga dapat memastikan diagnosis hemofilia dan dapat digunakan untuk diagnosis antenatal. Secara klinis, hemofilia A tidak dapat dibedakan dengan hemofilia B, oleh karena itu diperlukan pemeriksaan khusus F VIII dan IX.
Wanita pembawa sifat hemofilia A dapat diketahui dengan memeriksa kadar F VIII yang bisa di bawah normal, analisis mutasi gen hemofilia atau rasio F VIII dengan antigen faktor von Willebrand (FVIII/vWF:Ag ratio) yang kurang dari 1. Sedangkan wanita pembawa sifat hemofilia B dapat diketahui melalui aktivitas F IX yang dapat menurun atau pemeriksaan genetik.
Diagnosis banding hemofilia adalah penyakit von Willebrand, defisiensi faktor koagulasi lain seperti FV, FVII, FX, FXI, atau fibrinogen, atau kelainan trombosit seperti Glanzmann trombastenia.
Tatalaksana penderita hemofilia harus dilakukan secara komprehensif meliputi pemberian faktor pengganti yaitu F VIII untuk hemofilia A dan F IX untuk hemofilia B, perawatan dan rehabilitasi terutama bila ada sendi, edukasi dan dukungan psikososial bagi penderita dan keluarganya.
Bila terjadi perdarahan akut terutama daerah sendi, maka tindakan RICE (rest, ice, compression, elevation) segera dilakukan. Sendi yang mengalami perdarahan diistirahatkan dan diimobilisasi. Kompres dengan es atau handuk basah yang dingin, kemudian dilakukan penekanan atau pembebatan dan meninggikan daerah perdarahan. Penderita sebaiknya diberikan faktor pengganti dalam 2 jam setelah perdarahan.
Untuk hemofilia A diberikan konsentrat F VIII dengan dosis 0.5 x BB (kg) x kadar yang diinginkan (). F VIII diberikan tiap 12 jam sedangkan F IX diberikan tiap 24 jam untuk hemofilia B.4 Kadar F VIII atau IX yang diinginkan tergantung pada lokasi perdarahan dimana untuk perdarahan sendi, otot, mukosa mulut dan hidung kadar 30-50 diperlukan. Perdarahan saluran cerna, saluran kemih, daerah retroperitoneal dan susunan saraf pusat maupun trauma dan tindakan operasi dianjurkan kadar 60- 100%.
Lama pemberian tergantung pada beratnya perdarahan atau jenis tindakan. Untuk pencabutan gigi atau epistaksis, diberikan selama 2-5 hari, sedangkan operasi atau laserasi luas diberikan 7-14 hari. Untuk rehabilitasi seperti pada hemarthrosis dapat diberikan lebih lama lagi.
Kriopresipitat juga dapat diberikan untuk hemofilia A dimana satu kantung kriopresipitat mengandung sekitar 80 U F VIII. Demikian juga dengan obat antifibrinolitik seperti asam epsilon amino-kaproat atau asam traneksamat. Aspirin dan obat antiinflamasi non steroid harus dihindari karena dapat mengganggu hemostasis.
Profilaksis F VIII atau IX dapat diberikan secara kepada penderita hemofilia berat dengan tujuan mengurangi kejadian hemartrosis dan kecacatan sendi. WHO dan WFH merekomendasikan profilaksis primer dimulai pada usia 1- 2 tahun dan dilanjutkan seumur hidup. Profilaksis diberikan berdasarkan Protokol Malmö yang pertama kali dikembangkan di Swedia yaitu pemberian F VIII 20-40 U/kg selang sehari minimal 3 hari per minggu atau F IX 20-40 U/kg dua kali per minggu.
Untuk penderita hemofilia ringan dan sedang, desmopressin (1-deamino-8- arginine vasopressin, DDAVP) suatu anolog vasopressin dapat digunakan untuk meningkatkan kadar F VIII endogen ke dalam sirkulasi, namun tidak dianjurkan untuk hemofilia berat. Mekanisme kerja sampai saat ini masih belum jelas, diduga obat ini merangsang pengeluaran vWF dari tempat simpanannya (Weibel-Palade bodies) sehingga menstabilkan F VIII di plasma. DDAVP dapat diberikan secara intravena, subkutan atau intranasal.
Penderita hemofilia dianjurkan untuk berolah raga rutin, memakai peralatan pelindung yang sesuai untuk olahraga, menghindari olahraga berat atau kontak fisik. Berat badan harus dijaga terutama bila ada kelainan sendi karena berat badan yang berlebih memperberat arthritis. Kebersihan mulut dan gigi juga harus diperhatikan. Vaksinasi diberikan sebagaimana anak normal terutama terhadap hepatitis A dan B. Vaksin diberikan melalui jalur subkutan, bukan intramuskular. Pihak sekolah sebaiknya diberitahu bila seorang anak menderita hemofilia supaya dapat membantu penderita bila diperlukan.
Upaya mengetahui status pembawa sifat hemofilia dan konseling genetik merupakan hal yang terpadu dalam tatalaksana hemofilia. Konseling genetik perlu diberikan kepada penderita dan keluarga. Konseling meliputi penyakit hemofilia itu sendiri, terapi dan prognosis, pola keturunan, deteksi pembawa sifat dan implikasinya terhadap masa depan penderita dan pembawa sifat. Deteksi hemofilia pada janin dapat dilakukan terutama bila jenis mutasi gen sudah diketahui. Sampel dapat diperoleh melalui tindakan sampling villus khorionik atau amnionsintesis.
Sampai sekarang masih belum jelas mengapa perdarahan sendi atau hemarthrosis sering terjadi pada penderita hemofilia, namun diduga bahwa hal ini disebabkan oleh rendahnya ekspresi tissue factor di jaringan sinovial sehingga perdarahan mudah terjadi. Darah dan deposit besi dalam sendi mengiritasi sinovium dan merangsang reaksi inflamasi dalam sendi. Sinovitis kronis ini menyebabkan pertumbuhan jaringan sinovium yang penuh dengan pembuluh darah yang rapuh dan rawan terhadap perdarahan berikutnya, sehingga menciptakan suatu siklus setan. Sendi yang mengalami perdarahan berulang ini disebut sebagai sendi target. Hasil akhirnya adalah suatu arthropati hemofilik dimana sendi menjadi kaku, terjadi deformitas permanen, misalignment, perbedaan panjang anggota gerak serta hipotrofi otot yang berdekatan. Cacat sendi ini merupakan salah satu morbiditas penderita hemofilia yang utama.
Perdarahan intrakranial merupakan penyebab kematian utama penderita hemofilia. Studi di Inggris menunjukkan bahwa 34% kematian penderita hemofilia disebabkan oleh perdarahan ini, terutama di usia balita dimana 11 dari 13 kematian karena perdarahan intrakranial.19 Seumur hidupnya risiko perdarahan intrakranial pada seorang penderita hemofilia sebesar 2-8% dengan tingkat kematian 30%.
Perdarahan otot terutama terjadi di otot paha, betis, dinding perut bagian posterior dan bokong. Tekanan akibat perdarahan otot ini dapat mengakibatkan neuropati seperti neuropati nervus femoralis akibat perdarahan ileospoas. Nekrosis iskhemik dan kontraktur merupakan efek perdarahan otot lainnya.
Penularan penyakit seperti hepatitis C dan HIV melalui transfusi produk darah dan faktor pengganti merupakan masalah besar terutama pada tahun 1980 an. Upaya penapisan yang lebih baik saat ini telah sangat mengurangi risiko penularan tersebut, meskipun penularan Parvovirus B19 dan penyakti Creutzfeld- Jacob masih sulit dihindari. Kemajuan teknologi telah memungkinkan diproduksi faktor pengganti yang bebas dari risiko penularan penyakit tersebut dengan teknik rekombinan DNA.
Pembentukan antibodi atau inhibitor F VIII dapat timbul pada sekitar 20% penderita hemofilia A. Adanya inhibitor ini perlu dicurigai bila seorang penderita tidak menunjukkan penyembuhan yang diharapkan meski telah diberi faktor pengganti dengan dosis yang cukup. Dalam hal ini dosis F VIII harus dinaikkan atau diberikan F VIIa untuk memotong jalur koagulasi.
Menurut studi di Inggris, harapan hidup penderita hemofilia berat pada usia 35, 55 dan 75 tahun adalah 89%, 68% dan 23%, dengan median usia harapan hidup 63 tahun. Untuk penderita hemofilia sedang harapan hidup untuk kategori usia yang sama adalah 96%, 88% dan 49% dengan median usia harapan hidup 75 tahun. Sebagai perbandingan harapan hidup rerata pria di Inggris adalah 97%, 92% dan 59% dengan median usia harapan hidup 78 tahun.
Hemofilia merupakan penyakit perdarahan akibat gangguan koagulasi yang diturunkan. Penyakit perdarahan akibat gangguan koagulasi yang diturunkan meliputi: hemofilia A, hemofilia B, dan penyakit von Willebrand.
Dikenal 3 jenis hemofilia, yaitu: hemofilia A (defisiensi faktor VIII/anti hemophilic factor), hemofilia B (defisiensi faktor IX/Christmas factor), dan hemofilia C (defisiensi faktor XI).
Hemofilia A dan B merupakan penyakit perdarahan herediter berat yang paling sering, terjadi pada kira-kira 1: 5.000 laki-laki, sekitar 85% berupa hemofilia A dan 10-15% berupa hemofilia B. Hemofilia A dan B dapat terjadi pada semua golongan etnis. Gen faktor VIII dan IX terletak dekat ujung lengan panjang kromosom X oleh karena itu diturunkan secara X-linked traits/recessive, sehingga biasanya perempuan sebagai pembawa sifat sedangkan laki-laki sebagai penderita. Defisiensi faktor XI merupakan defisiensi otosomal yang berhubungan dengan gejala perdarahan ringan- sedang.
Penyakit von Willebrand merupakan penyakit perdarahan herediter yang paling sering dengan angka kejadian sekitar 1-2% populasi, diturunkan secara otosomal, sehingga dapat terjadi pada perempuan dan laki-laki, disebabkan karena abnormalitas faktor von Willebrand (FVW) baik kuantitatif dan atau kualitatif.
Klasifikasi beratnya hemofilia bergantung pada kadar faktor VIII atau faktor IX dalam plasma. Diklasifikasikan hemofilia berat bila kadar kadar F.VIII atau F.IX kurang dari 1%, hemofilia sedang bila kadarnya 1-5% dan ringan bila kadarnya 5-30%. Kadar hemostatik untuk F.VIII adalah > 30-40%, dan untuk F.IX adalah >25-30%.
Klasifikasi penyakit von Willebrand meliputi: tipe 1 (defisiensi kuantitatif parsial FVW), merupakan tipe yang paling sering (sekitar 85%); tipe 2 (karena kualitas yang abnormal), terdiri dari tipe 2A, 2B, 2N, 2M; dan tipe 3 yang merupakan defisiensi FVW total. Tipe 1, 2A dan 2B diturunkan secara autosomal dominant, sedangkan tipe 2N dan 3 merupakan keadaan yang resesif.
Dalam kaskade koagulasi faktor VIII dan IX bersama dengan fosfololipid dan kalsium berperan dalam mengaktivasi faktor X melalui jalur intrinsik. Defisiensi faktor-faktor tersebut akan menyebabkan gangguan pembentukan bekuan fibrin. Sedangkan FVW berperan penting dalam memulai proses hemostasis dengan cara meningkatkan adhesi dan agregasi trombosit.
Pertama, FVW memediasi adhesi trombosit dengan endotel dengan cara berikatan dengan reseptor trombosit glikoprotein Ib (GPIb), kemudian memfasilitasi interaksi dengan GPIIb sehingga terjadi agregasi trombosit.
Kedua, FVW berperan sebagai pembawa protein faktor VIII sehingga bila kadar FVW rendah akan mengakibatkan kadar faktor VIII juga rendah (defisiensi
F.VIII sekunder). Mutasi pada beberapa lokus yang berbeda menyebabkan timbulnya berbagai varian penyakit ini.
Gambaran klinis hemofilia A dan B sulit dibedakan. Manifestasi klinis perdarahan pada hemofilia A dan B sejalan dengan derajat defisiensinya. Perdarahan yang umum dijumpai adalah mudah memar, perdarahan oral khususnya perdarahan gusi, hemartrosis dan hematoma yang terjadi secara spontan atau setelah adanya trauma. Perdarahan yang terjadi pada penyakit von Willebrand dapat berupa perdarahan ringan sampai berat, biasanya berupa perdarahan mukokutan seperti memar yang hebat, epistaksis, menoragi, adanya perdarahan yang memanjang pada luka kecil, perdarahan yang berlebihan setelah trauma atau cabut gigi.
Diagnosis ditegakkan berdasarkan atas anamnesis, pemeriksaan fisik dan laboratorium. Pada anamnesis didapatkan adanya keluhan perdarahan spontan atau karena trauma, dicari riwayat keluarga dengan keluhan yang sama meskipun pada sekitar 20-30% tidak didapatkan riwayat keluarga (terjadi karena adanya mutasi spontan), pada pemeriksaan fisik dicari tanda- tanda perdarahan, dan pada pemeriksaan laboratorium didapatkan masa tromboplastin parsial teraktifasi (aPTT) memanjang.
Diagnosis pasti ditegakkan berdasarkan hasil pemeriksaan laboratorium dengan pemeriksaan kadar faktor VIII untuk hemofilia A dan kadar faktor IX untuk hemofilia B. Diagnosis pasti penyakit von Willebrand ditegakkan berdasarkan anamnesis yang sugestif untuk penyakit ini dibantu dengan pemeriksaan laboratorium spesifik. Umumnya didapatkan waktu perdarahan dan aPTT yang memanjang. Hasil normal pada tes skrining belum menyingkirkan diagnosis penyakit ini. Diagnosis pasti ditegakkan berdasarkan hasil pemeriksaan kadar F.VIII, antigen FVW (VWF:Ag), aktivitas FVW (VWF R:Co) dan VWF multimers.
Pengobatan pasien hemofilia harus dilakukan secara komprehensif, selain mengganti faktor pembekuan yang kurang, perawatan serta rehabilitasi penting dilakukan, diperlukan juga edukasi bagi pasien dan keluarganya. Bila terjadi perdarahan akut pada hemofilia maka yang harus dilakukan pertama ialah tindakan imobilisasi, kompres es. Penekanan atau pembebatan serta meninggikan daerah yang mengalami perdarahan juga perlu dilakukan.
Dalam 2 jam setelah perdarahan, pasien hemofilia sudah harus mendapat faktor pembekuan yang diperlukan. Untuk hemofilia A diberikan konsentrat F. VIII dengan dosis (unit): unit/dL () kenaikan kadar yang diinginkan X BB (kg) X 0,5, dapat juga dengan dosis empiris yaitu untuk F. VIII 20-25 U/kg setiap 12 jam. Untuk hemofilia B diberikan konsentrat F. IX dengan dosis (unit): unit/dL () kenaikan kadar yang diinginkan X BB (kg), dapat juga diberikan dosis empiris 40-50 U/kg setiap
24 jam. Keduanya diawali dengan dosis muatan (loading dose) dua kali dosis rumatan.
Selanjutnya dilakukan evaluasi terhadap respon terapi. Bila konsentrat F.VIII tidak tersedia dapat diberikan kriopresipitat, sedangkan bila konsentrat F. IX tidak tersedia dapat diberikan FFP.
Pemberian komponen darah pada penyakit von Willebrand diperlukan untuk tindakan operasi yang cukup besar atau untuk mengatasi perdarahan berat yang mengancam jiwa.
Komplikasi penyakit hemofilia meliputi artropati kronis, timbulnya inhibitor karena terbentuknya antibodi baik untuk faktor VIII maupun faktor IX, dan risiko penyakit yang ditularkan lewat transfusi.
Inhibitor dilaporkan terjadi pada sekitar 25-35% pasien hemofilia A, pada hemofilia B angka kejadian inhibitor jauh lebih rendah (1-3%). Untuk mengatasi inhibitor diberikan terapi dengan cara induksi toleransi imun dengan menaikkan dosis faktor VIII atau dengan pemberian faktor VIIa rekombinan atau activated prothrombin complex concentrate (APCC) untuk memotong jalur koagulasi.
Hal lain yang cukup penting adalah pencegahan terjadinya perdarahan dengan menghindari trauma. Di negara maju pemberian profilaksis konsentrat F.VIII atau F. IX dilakukan secara rutin, sedangkan di negara berkembang hal ini belum dilakukan karena memerlukan biaya yang sangat mahal.
Prognosis ditentukan oleh derajat beratnya penyakit, pemberian terapi substitusi yang adekuat serta ada tidaknya penyulit.
Studi kasus 1 (Hemofilia A)
Seorang anak laki-laki berusia 2 tahun dibawa ibunya ke poliklinik anak RS dengan keluhan utama lutut kanannya bengkak. Keluhan muncul sejak 2 hari yang lalu, kemudian penderita tampak kesakitan sehingga tidak dapat berjalan. Ibunya mengatakan bahwa sebelumnya diketahui ada memar pada lengan dan tungkainya. Keluhan tidak didahului oleh adanya trauma. Penderita tampak sehat, tidak ada kejang, penurunan kesadaran ataupun pucat. Penderita baru pertamakali mengalami keluhan seperti ini. Tidak ada riwayat keluarga yang memiliki keluhan serupa. Penderita belum mendapat pengobatan.
Penilaian:
Jawaban:
Identifikasi masalah:
Melakukan pemeriksaan fisik dan perawatan awal berupa imobilisasi, kompres es, pembebatan sendi lutut, meninggikan tempat yang mengalami perdarahan.
Hasil penilaian yang ditemukan:
Diagnosis:
Berdasarkan hasil temuan tersebut apakah diagnosis kerja dan diagnosis banding penyakit pasien ini? Sebutkan alasannya!
Jawaban:
Diagnosis kerja: Hemofilia A
(Klinis ada manifestasi perdarahan berupa ekimosis dan hemartrosis. Gejala ini sesuai dengan gejala penyakit perdarahan akibat gangguan koagulasi. Adanya hemartrosis mengindikasikan penyakit hemofilia. Difikirkan hemofilia A karena merupakan jenis hemofilia yang paling sering).
Diagnosis banding: Hemofilia B
(Gejala klinis hemofilia A dan B sulit dibedakan. Diagnosis pasti ditegakkan berdasarkan hasil pemeriksaan laboratorium.)
Tata laksana (intervensi dan perencanaan):
Berdasarkan diagnosis tersebut bagaimanakah tata laksana awal pasien ini? Jawaban:
Bila dari hasil pemeriksaan laboratorium didapatkan darah rutin dalam batas normal, BT dan PT normal, aPTT memanjang, maka diagnosis pasien ini adalah Tersangka Hemofilia A (berat).
Bagaimanakah tata laksana selanjutnya untuk pasien ini?
Jawaban:
Pemberian terapi substitusi berupa konsentrat F. VIII dengan dosis: Hari ke-1: 0,5 x 10 x 50U= 250U (= 1 vial konsentrat F.VIII @250U)
Evaluasi respon terapi. Bila perdarahan berat (dan sendi masih terasa nyeri), terapi dilanjutkan setiap hari sampai fungsi sendi kembali normal dan pertimbangkan terapi tambahan selang sehari selama 1 minggu.
Bila konsentrat faktor VIII tidak tersedia diberikan kriopresispitat sebanyak: 10 x 50U= 500U
= 7 unit kriopresipitat (1 unit/bag kriopresipitat mengandung lebih kurang 70-80U F.VIII).
Penyuluhan: pencegahan trauma, edukasi untuk orang tua,
Rujuk untuk diagnosis pasti dan konseling genetik.
Bagaimanakah pemantauan pasien ini?
Jawaban:
Penilaian ulang
Apakah yang harus dipantau dalam tindak lanjut pasien selanjutnya ?
Jawaban:
Studi kasus 2 (Hemofilia dengan komplikasi)
Seorang anak laki-laki berusia 5 tahun dibawa ibunya ke poliklinik anak RS Hasan dengan keluhan utama nyeri lutut kanan. Keluhan muncul sejak 2 hari yang lalu, sebelumnya sejak 1 minggu lutut kanan sudah bengkak yang makin lama makin bertambah bengkak. Ibunya mengatakan bahwa sebelumnya diketahui ada memar pada lengan dan tungkainya. Keluhan tidak didahului oleh adanya trauma. Penderita tampak sehat, tidak ada kejang, penurunan kesadaran ataupun pucat. Penderita sudah sering mengalami keluhan serupa, dibawa berobat ke RS terdekat bila penderita mengeluh nyeri dan pernah mendapatkan transfusi darah berwarna kuning. Tidak ada riwayat keluarga yang memiliki keluhan serupa.
Penilaian:
Jawaban:
Identifikasi masalah:
Melakukan pemeriksaan fisik dan perawatan awal berupa imobilisasi, kompres es, pembebatan sendi lutut, meninggikan tempat yang mengalami perdarahan.
Hasil penilaian yang ditemukan:
Diagnosis:
Berdasarkan hasil temuan tersebut apakah diagnosis kerja dan diagnosis banding penyakit pasien ini? Sebutkan alasannya!
Jawaban:
Diagnosis kerja: Hemofilia A + artropati hemofilik
(Klinis ada manifestasi perdarahan berupa hemartrosis berulang dan ekimosis. Gejala ini sesuai dengan gejala penyakit perdarahan akibat gangguan koagulasi. Adanya hemartrosis mengindikasikan penyakit hemofilia. Difikirkan hemofilia A karena merupakan bentuk hemofilia tersering dan pasien diduga pernah mendapat kriopresipitat?).
Diagnosis banding: Hemofilia B
(Gejala klinis hemofilia A dan B sulit dibedakan. Difikirkan hemofilia B, karena mungkin transfusi yang pernah diperoleh pasien adalah FFP. Diagnosis pasti ditegakkan berdasarkan hasil pemeriksaan laboratorium.)
Tata laksana (intervensi dan perencanaan):
Berdasarkan diagnosis tersebut bagaimanakah tata laksana awal pasien ini?
Jawaban:
Bila hasil pemeriksaan laboratorium didapatkan darah rutin dalam batas normal, BT dan PT normal, aPTT memanjang, foto lutut kanan menunjukkan artropati maka diagnosis pasien ini adalah Tersangka Hemofilia A (berat)+ artropati hemofilik.
Bagaimanakah tata laksana selanjutnya untuk pasien ini?
Jawaban:
Pemberian terapi substitusi berupa konsentrat F. VIII dengan dosis: Hari ke-1: 0,5 x 20 x 50U= 500U (= 1 vial konsentrat F.VIII @250U)
Evaluasi respon terapi. Bila perdarahan berat dan sendi masih terasa nyeri, terapi dilanjutkan setiap hari sampai fungsi sendi kembali normal dan pertimbangkan terapi tambahan selang sehari selama 1 minggu.
Bila konsentrat faktor tidak tersedia dapat diberikan kriopresispitat sebanyak: 20x 50U= 1000U= 14 unit kriopresipitat (1 unit/bag kriopresipitat mengandung lebih kurang
70-80U F.VIII). Evaluasi selanjutnya sama dengan di atas.
Penyuluhan: pencegahan trauma, edukasi untuk orang tua, konseling genetik
Merujuk pasien ke Pusat Pelayanan Hemofilia (untuk rencana pemberian terapi profilaksis, fisioterapi ( Rehabilitasi medik), tata laksana bedah/sinovektomi (Bedah Ortopedi), terapi substitusi untuk sebelum, selama dan sesudah operasi, konseling genetik.
Bagaimanakah pemantauan pasien ini?
Jawaban:
Penilaian ulang
Apakah yang harus dipantau dalam tindak lanjut pasien selanjutnya?
Jawaban:
Studi kasus 3 (Penyakit von Willebrand)
Seorang anak perempuan berusia 7 tahun dibawa ibunya ke poliklinik anak RS dengan keluhan utama perdarahan hidung (mimisan). Keluhan muncul sejak 3 hari yang lalu, kemudian perdarahan makin bertambah meskipun sudah dilakukan penekanan hidung. Ibunya mengatakan bahwa penderita sudah sering mengalami keluhan seperti ini, tetapi biasanya perdarahan dapat berhenti sendiri. Penderita juga sering mengalami biru-biru pada lengan dan tungkai bawahnya yang kemudian dapat menghilang. Keluhan tidak didahului oleh adanya trauma. Penderita tampak sehat, tidak tampak pucat. Penderita baru pertamakali mengalami keluhan seperti ini. Tidak ada riwayat keluarga yang memiliki keluhan serupa. Penderita belum pernah mendapat pengobatan.
Penilaian:
Jawaban:
Identifikasi masalah:
Melakukan pemeriksaan fisik dan perawatan awal berupa penekanan (tampon hidung) Hasil penilaian yang ditemukan:
Diagnosis:
Berdasarkan hasil temuan tersebut apakah diagnosis kerja dan diagnosis banding penyakit pasien ini? Sebutkan alasannya!
Jawaban:
Diagnosis kerja: Penyakit von Willebrand (tipe 1)
(Klinis seorang anak perempuan dengan manifestasi perdarahan mukokutan berupa epistaksis dan ekimosis. Gejala ini sesuai dengan gejala penyakit perdarahan akibat gangguan hemostasis karena abnormalitas FVW. Difikirkan penyakit von Willebrand karena dari anamnesis sugestif kearah penyakit ini (perempuan, perdarahan mukokutan) dan angka kejadiannya cukup tinggi. Difikirkan tipe 1 karena merupakan tipe tersering.
Diagnosis banding: ITP Trombositopati
(Gejala klinis kelainan hemostasis akibat kelainan vaskular dan trombosit sering serupa. Manifestasi klinis kelainan trombosit baik jumlah maupun fungsi sama. Diagnosis pasti ditegakkan berdasarkan hasil pemeriksaan laboratorium.)
Tata laksana (intervensi dan perencanaan):
Berdasarkan diagnosis tersebut bagaimanakah tata laksana awal pasien ini?
Jawaban:
Bila dari hasil pemeriksaan laboratorium didapatkan kadar trombosit, BT dan aPTT normal, belum dapat menyingkirkan diagnosis penyakit Von Willebrand.
Umumnya kadar trombosit normal, BT memanjang, aPTT dapat normal atau memanjang.
Bagaimanakah tata laksana selanjutnya untuk pasien ini?
Jawaban:
Bagaimanakah pemantauan pasien ini?
Penilaian ulang
Apakah yang harus dipantau dalam tindak lanjut pasien selanjutnya ?
Jawaban: